Каналопатии
Каналопатии – генетически обусловленные заболевания, связанные с нарушением структуры и функции ионных каналов.
Каналопатии имеют преимущественно наследственный характер и являются первичными, и в этом случае в качестве причин рассматриваются исключительно мутации в генах, кодирующих тот или иной элемент ионного канала. Также каналопатии могут иметь вторичный характер, что проявляется в результате воздействия аутоиммунных, метаболических, токсических процессов, а также под воздействием ряда лекарственных препаратов.
Деление каналопатий на первичные и вторичные достаточно условно, так как в последнее время все чаще упоминается о том, что пациенты, у которых выявлена вторичная каналопатия, с высокой вероятностью имеют первичный генетический дефект, проявшийся лишь под воздействием вышеуказанных факторов.
К сердечным каналопатиям в настоящее время принято относить:
- Синдром удлиненного интервала QT (LQTS)
- Синдром укороченного интервала QT (SQTS)
- Синдром Бругада (BrS)
- Катехоламинергическая полиморфная желудочковая тахикардия (КПЖТ, CPVT).
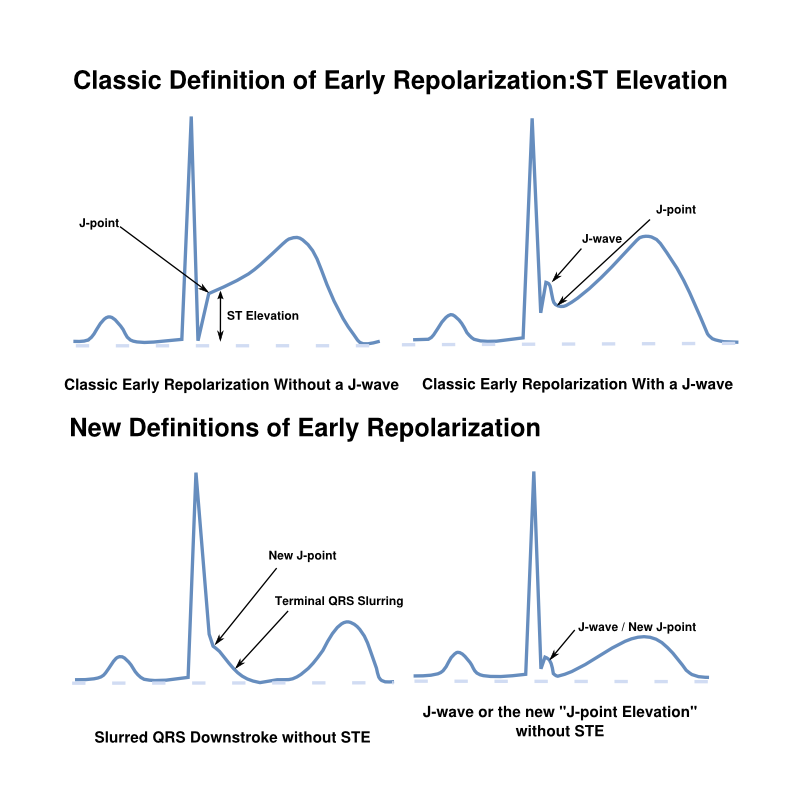
- Синдром ранней реполяризации желудочков (СРРЖ, ERS)
Все пять сердечных каналопатий имеют ряд общих особенностей:
- Специфические для каждой каналопатии изменения на ЭКГ.
- Неспецифическая клиническая картина в виде обмороков и внезапной смерти, возникающих на фоне желудочковых аритмий, причем внезапная смерть может стать одновременно и дебютом, и драматическим финалом заболевания.
- Генетическая обусловленность и преимущественно аутосомно-доминантный путь наследования.
- Анатомически здоровое сердце.
Первичная диагностика сердечных каналопатий, а также их дифференциальная диагностика основана в первую очередь на знании особенностей электрокардиографических фенотипов, характерных для каждого из этих заболеваний:
- синдром удлиненного или укороченного интервала QT на ЭКГ выглядит, соответственно, либо удлинением, либо укорочением корригированного интервала QT сверх установленных значений (QTc ≥ 480 или ≤ 340 мс), с учетом ряда нюансов.
- для диагностики синдрома Бругада необходимо наличие специфического ЭКГ–паттерна (1 ЭКГ-тип синдрома Бругада) в правых грудных отведениях, который возникает либо спонтанно, либо после медикаментозного провокационного теста одним из антиаритмических препаратов IA и IC классов (аймалин, прокаинамид, флекаинид).
Изменения ЭКГ именно в правых грудных отведениях обусловлены аномальной электрофизиологической активностью эпикарда правого желудочка в области выносящего тракта, что характерно только для синдрома Бругада.
- КПЖТ можно диагностировать лишь непосредственно в момент приступа аритмии, для которой будут характерны все ЭКГ- признаки полиморфной желудочковой тахикардии. Характерно, что приступ возникает вследствие острой активации симпатической нервной системы (на фоне физической или эмоциональной нагрузки).
- СРРЖ характеризуется наличием специфической J–волны или точки-J на нисходящей части комплекса QRS не менее чем в двух смежных отведениях, независимо от того, имеется ли при этом элевация сегмента ST или нет. При этом максимально неблагоприятными в отношении риска внезапной смерти являются изменения в нижних (II, III, aVF) отведениях ЭКГ.
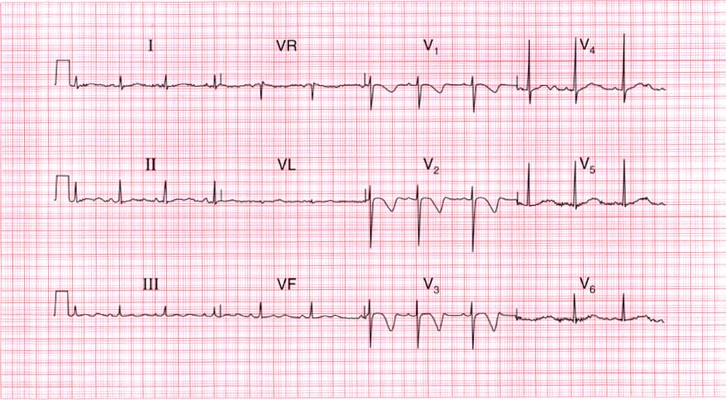
Сердечные каналопатии довольно редко выявляются в обычной клинической практике, но при этом являются одной из основных причин внезапной сердечной смерти (ВСС) у детей и лиц молодого возраста, не имеющих органических и структурных заболеваний сердца.
Механизм развития внезапной смерти обусловлен развитием фатальных желудочковых аритмий, а развитие желудочковых аритмий обусловлено изменением потенциала действия кардиомиоцитов, который формируется за счет работы единого суперсемейства потенциал-активируемых натриевых, кальциевых и калиевых ионных каналов, и изменяется по-разному при каждой каналопатии.
Диагностику сердечных каналопатий существенно затрудняет разнообразие клинических вариантов их течения: от скрытых форм до классических, которые в свою очередь могут проявляться либо только изменениями на ЭКГ (постоянными или преходящими), либо только обмороками, либо и тем и другим. Интересно, что синдром Бругада чаще имеет скрытый вариант течения, в то время как при синдроме удлиненного интервала QT чаще встречаются классические явные варианты. Понятно, что в случае скрытых форм и при отсутствии подтверждающего ДНК-анализа, диагноз поставить невозможно.
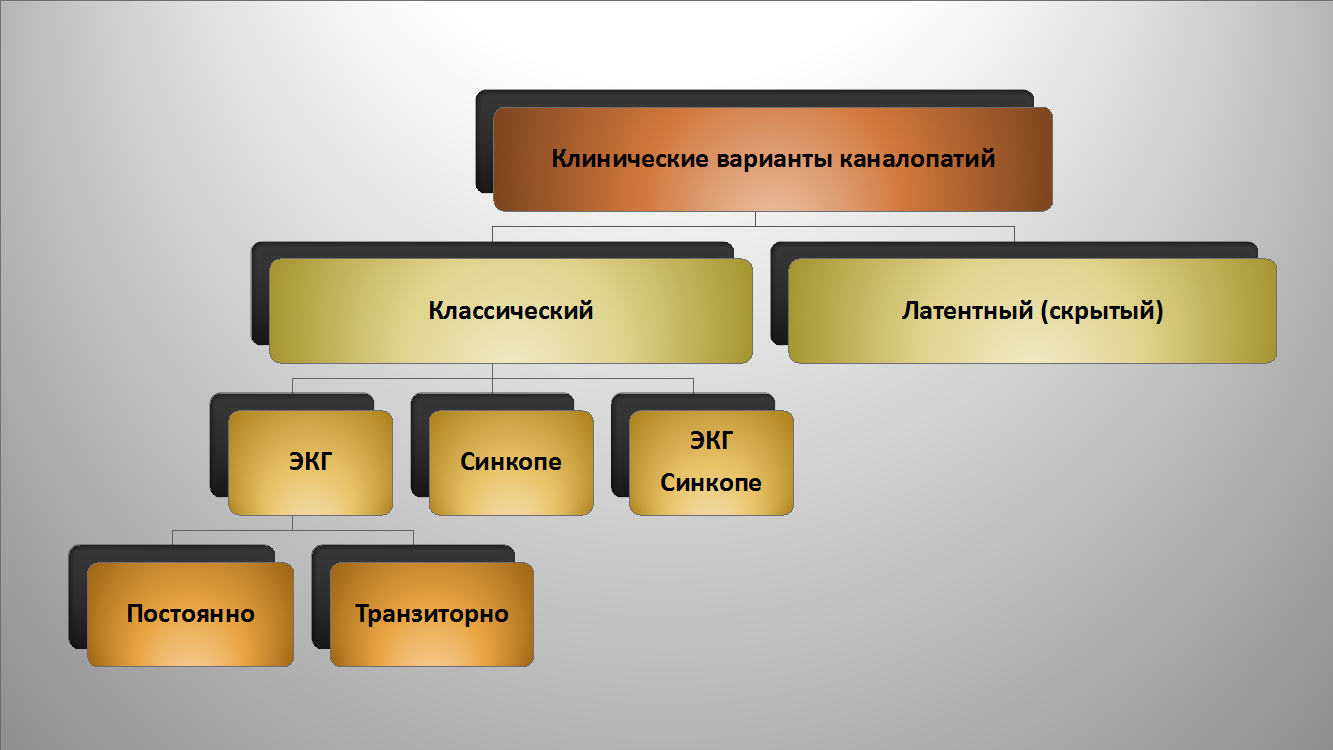
Кроме клинического разнообразия, сердечные каналопатии еще и достаточно гетерогенны генетически. Каждая каналопатия ассоциируется с мутациями в генах, кодирующих непосредственно калиевые, натриевые и кальциевые каналы, а также генах, кодирующих либо связывающие белки, либо рецепторы или ферменты, играющие важную роль в строении и работе ионных каналов. На сегодняшний день суммарно насчитывается около 40 генотипов сердечных каналопатий, но хорошо известно, что достаточно большой процент генотипов остается до сих пор неуловимым (Например, 75% случаев синдрома Бругада являются неуловимыми).
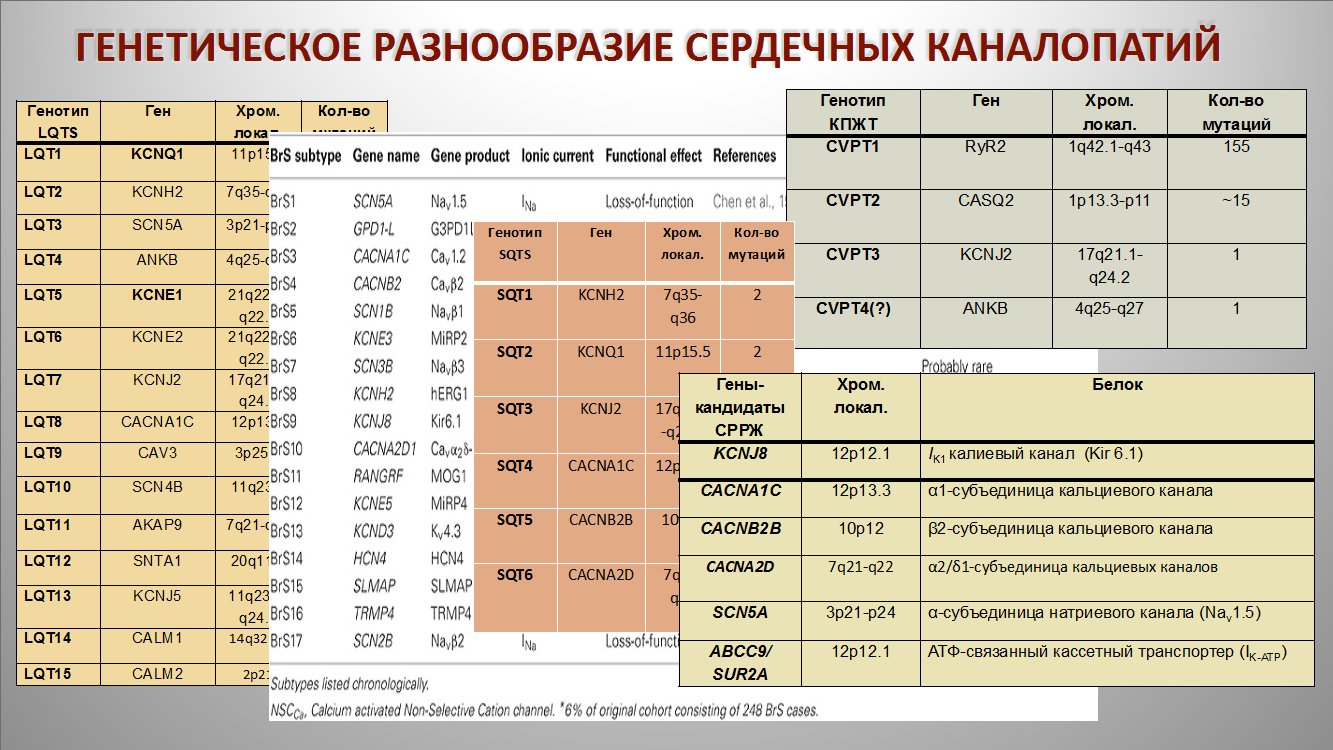
Кроме генетического разнообразия сердечные каналопатии имеют еще целый ряд генетических особенностей:
- Низкая пенетрантность
- Корреляция генотип/фенотип слабо выражена и имеется у малого количества генотипов
- Полилокусные (один фенотип – несколько генов)
- Имеются аллельные серии заболеваний (один ген – несколько фенотипов)
- Мутации de novo
- Генетическая неуловимость (20-25% для LQTS и 75% для BrS)
- Бессимптомные носители имеют меньший риск ВСС, но он существенно повышается при лекарственном индуцировании.
Учитывая сложности клинической и генетической диагностики, необходимо использовать следующий упрощенный алгоритм выявления сердечных каналопатий:
- Оценка изменений на ЭКГ, включая суточное мониторирование ЭКГ.
- Исключить вторичные причины изменений на ЭКГ.
- При необходимости провокационная проба: лекарственная (при подозрении на синдром Бругада) или с физической нагрузкой (при подозрении на КПЖТ).
- Тщательный анамнез заболевания (возраст и характеристика дебюта, частота симптомов и т.п.)
- Сбор семейного анамнеза.
- Семейное ЭКГ обследование.
- Генетическое тестирование (объем ДНК-диагностики выбирается в индивидуальном порядке в зависимости от конкретных целей).
Генетический анализ нужен не только для уточнения генетической формы заболевания и подтверждения диагноза, но и для правильной оценки риска ВСС, подбора индивидуальной (в ряде случаев гено-специфической) тактики лечения, проведения каскадного скрининга семьи и медико-генетического консультирования, что особенно важно в случае отсутствия структурных изменений и при бессимптомном течении заболевания.
В тоже время необходимо понимать, что ДНК-диагностика в отношении сердечных каналопатий обладает лишь подтверждающей силой, поскольку отрицательный генетический анализ при наличии положительной клинической картины никак не позволяет исключить заболевание.
Таким образом, на сегодняшний день принято полагать, что клиническая симптоматика и данные анамнеза являются основными критериями в постановке диагноза той или иной сердечной каналопатии, а генетический анализ - лишь вспомогательное действие, но не менее важное для дальнейшего понимания патогенеза этих сложных редких заболеваний.
Артериальная гипертензия
Считается, что генетические факторы определяют 30%-50% вариаций артериального давления. Эссенциальная артериальная гипертензия (АГ)— крайне неоднородное состояние с многофакторной этиологией. Несколько исследований по изучению всего генома и их мета-анализы указывают на существование, в общей сложности, 29 однонуклеотидных полиморфизмов, ассоциированных с систолическим и/диастолическим АД. Эти данные, возможно, могут быть использованы в шкалах оценки сердечно-сосудистого риска.
В клинической практике важно обнаружить или исключить редкие, моногенные формы наследственной АГ. К ним относятся, в частности, синдром Лиддла, патология амилорид-чувствительных эпителиальных натриевых каналов, синдром кажущейся избыточности минералокортикоидной активности и гиперальдостеронизм, корригируемый глюкокортикоидами. Генетическое исследование и обнаружение мутантного гена позволяет в таких случаях выявить причину АГ и в ряде случаев провести патогенетическую терапию.
Большинство форм генетически обусловленных артериальных гипертоний характеризуется синдромом гипертонии – гипокалиемии и связаны с наличием патологических вариантов генов, связанных с функционированием системы альдостерона.
Первичный гиперальдостеронизм
Первичный гиперальдостеронизм (ПГА) - собирательный диагноз, включающий в себя ряд различных состояний, характеризующихся повышением уровня альдостерона, который относительно автономен от ренин-ангиотензиновой системы и не снижается при натриевой нагрузке.
Повышение уровня альдостерона является причиной множественных расстройств, включая снижение активности ренина плазмы крови, повышение артериального давления (АД), задержку натрия, и ускоренное выделения калия, что приводит к гипокалиемии. Среди различных клинических форм ПГА аденома надпочечника, односторонняя или двусторонняя надпочечниковая гиперплазия, в том числе и семейные формы, в редких случаях — наследственно обусловленный глюкокортикоид-зависимый альдостеронизм.
Семейный первичный альдостеронизм I типа (глюкокортикоид-зависимый альдостеронизм)
Заболевание характеризуется рано дебютирующей гипертонией, увеличением активности альдостерона, гиперпродукцией гормонов коры надпочечников (18-оксокортизола и 18-гидроксикортизола). Для больных характерна высокая активность альдостерона, низкая активность ренина крови, двусторонняя гиперплазия коры надпочечников. Повышение АД оказывается более тяжелым у мужчин. Дебют АГ приходится на 1-2 десятилетие жизни (чаще в возрасте 10-20 лет). Частым осложнением заболевания считается развитие цереброваскулярных осложнений в молодом возрасте. АГ оказывается резистентной к стандартной терапии. Практически всегда семейный анамнез оказывается отягощенным по случаям ранней АГ и инсультов 5.
Согласно Европейскому консенсусу показаниями к генетическому скринингу семейного гиперальдостеронизма I типа являются:
- выявленный первичный альдостеронизм в возрасте до 20 лет (с или без гипокалиемии)
- выявленный альдостеронизм в возрасте до 20 лет в сочетании с отягощенным семейным анамнезом
- выявленный альдостеронизм и инсульт в возрасте до 40 лет.
Псевдоальдостеронизм
Синдром Лиддла моногенное аутосомно-доминантное заболевание (MIM177220), характеризующееся рано дебютирующей резистентной к обычной терапии артериальной гипертонией и гипокалиемией. В тяжелых случаях заболевание может сопровождаться метаболическим алкалозом. Впервые заболевание было описано Лиддлом в 1963 году. Причиной развития заболевания является мутации в генах SCNN1B или SCNN1G, кодирующих бета- или гамма - субъединицу эпителиального натриевого канала (ENaC) дистальных почечных канальцах и собирательных трубочках.
Для скрининга синдрома Лиддла у взрослых предложены следующие критерии
- Натрий крови выше 140 ммоль/л в двух и более анализах
- Калий крови ниже 3,5 ммоль/л в двух и более анализах
- Бикарбонаты крови выше 28 ммоль/л в двух и более анализах
- Необходимо наличие по крайней мере 2 признаков.
Псевдоальдостеронизм II типа (синдром Гордона)
Cиндром гиперкалиемии – гипертонии. Тип наследования – аутосомно-доминантный. Выделяют несколько подтипов, описанных у разных семей.
Болеют как дети, так и взрослые. Популяционная частота неизвестна. Описано более 80 семей и более 180 больных.
Заболевание характеризуется гиперкалиемией при сохранённой скорости клубочковой фильтрации, гипертонией и высокой эффективностью тиазидных диуретиков для коррекции АГ. Также характерным является незначительное повышение уровня магния крови, метаболический ацидоз, сниженная активность ренина крови. Формализованных диагностических критериев нет.
Характерно повышение АД в подростковом или детском возрасте сопровождающееся
- Гиперкалиемией при сохранной СКФ
- Уровнем калия крови от 5.0-6.0 ммоль/л до 8.0 ммоль/л
Указанные изменения могут быть выявлены у больных любого возраста
- Метаболический ацидоз – бикарбонаты плазмы 14 -24 ммоль/л
- Гиперхлоремия – хлориды 105 - 117 ммоль/л
- Снижение активности ренина
- Вариабельный уровень альдостерона со склонностью к снижению
- Кальций крови и парат-гормон в норме, однако возможна гиперкальциурия.
Семейный анамнез и консультирование – рекомендуется обследование родственников 1 степени родства. Отсутствие случаев в семье не исключает наличие заболеваний у пробандов.
Подтверждение заболевания идентификация гетерозиготного патогенного варианта в генах CUL3, WNK1, или WNK4 или гетеро – или гомозиготного варианта в гене KLHL3.
Метаболические нарушения и уровень артериального давления успешно корректируется назначением тиазидных диуретиков в стандартных дозах с контролем уровня электролитов крови. При необходимости доза тиазидов корректируется с учетом уровня электролитов.
Больным рекомендуется ограничивать потребление натрия и калия с пищей.
Синдром Геллера
Аутосомно-доминантная рано дебютирующая гипертония, течение которой ухудшается при беременности
Впервые заболевание описано Геллером в 2000 году. При обследовании 75 больных с тяжелой, рано дебютировавшей АГ, был выявлен пациент 15 лет, с тяжелой АГ, низким уровнем ренина и альдостерона и отсутствием каких-либо причин, вызывающих повышение АД. При проведении генетического исследования выявлена мутация Leu 810 Ser в гене минералокортикоидного рецептора (NR3C2). Описанная мутация делает МР чувствительным к прогестерону и другим частичным агонистам МР. Применение блокаторов альдостероновых рецепторов также может утяжелить течение АГ. Во время беременности при нарастании уровня прогестерона он также становится стимулятором МР. Развивается значимое повышение АД, гипокалиемия и низком или неопределяемом уровне альдостерона. Повышение АД особенно выражено во 2-3 триместре беременности, может сопровождаться развитием отечного синдрома, преэклампсии и эклампсии. АГ оказывается резистентной к обычному лечению.
Синдром кажущегося избытка минералокортикоидов
Клиническая картина – заболевание характеризуется тяжелой АГ, низким уровнем ренина и альдостерона, гипокалиемией, гипернатриемией, полиурией, метаболическим алкалозом. Заболевание дебютирует у детей в возрасте до 10 лет. При обследовании родителей часто выявляется АГ с низким уровнем ренина, но без выраженного изменения уровня электролитов и с нормальным уровнем альдостерона.
Характер наследования – аутосомно-рецессивный.
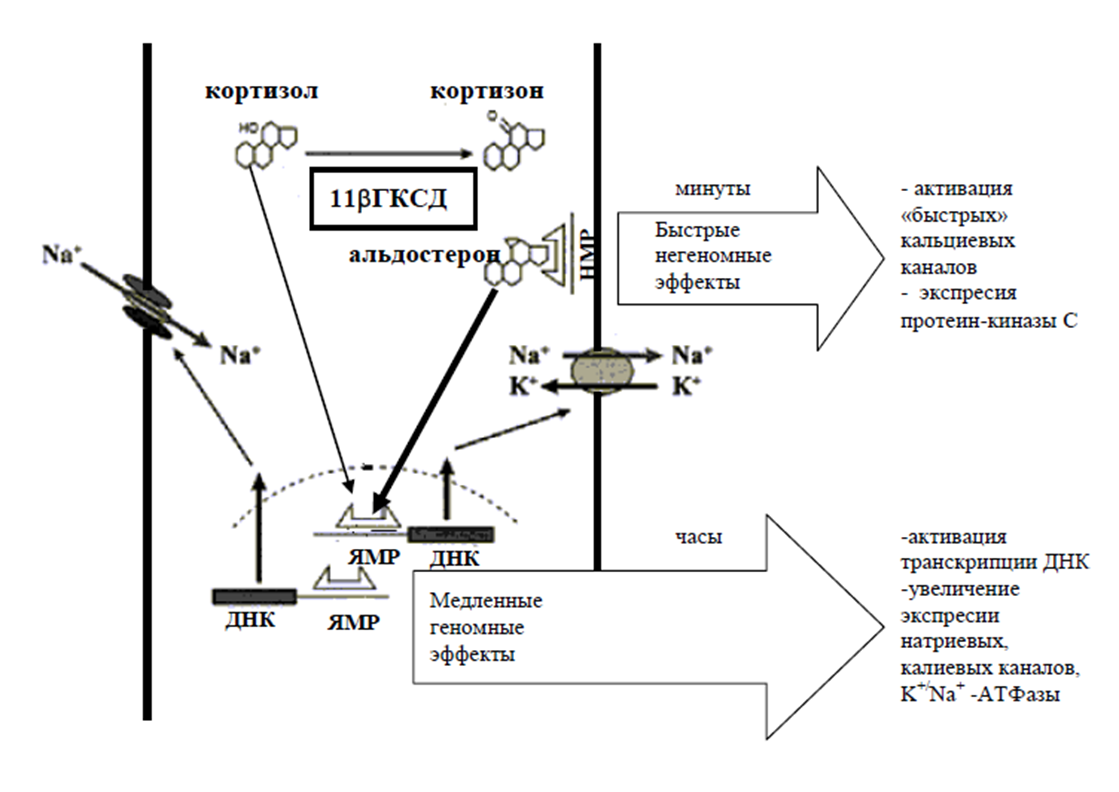
Заболевание связано с мутацией в гене 11-бета гидроскистероид-дегидрогеназы 2 типа (HSD11B2). Ген картирован в хромосоме 16q22.
Функция фермента – конверсия кортизола в кортизон. При наличии мутаций функция фермента снижается, создается избыток кортизола, который обладает большим сродством к минералокортикоидным рецепторам, чем альдостерон. Синтез альдостерона при этом подавляется и развивается клиническая картина псевдоальдостеронизма.
Описаны мутации Ala221Gly, ARG208CYS, ARG213CYS, ARG337CYS, ARG279CYS, PRO227LEU и некоторые другие, вызывающие развитие заболевания.
Фибромускулярная дисплазия
Идиопатическое сегментарное не атеросклеротическое заболевание, поражающее мышечный слой артерий малого и среднего калибра и приводящее к их стенозированию. Описано три основных типа – интимальный (5%), медиальный (85%) и перимедиальный (10%) типы. Для фибромускулярной дисплазии характерным считается появление микроаневризм с появлением стенозирования между аневризмами. Фибромускулярная дисплазия считается редким заболеванием с распространённостью 1:200 000 населения, однако данные анализа аутопсийного материала донорских почек показал, что распространенность ФМД может достигать 4%, причем большинство случает остается бессимптомными.
Наиболее часто ФМД поражает почечные артерии, однако возможны и другие локализации:
- Почечные артерии (60-75%)
- Артерии головы и шеи (25-30%)
- Висцеральные сосуды (9%)
- Артерии конечностей (5%)
- Легочные и коронарные сосуды - редко
- Множественные локализации – до 28% больных
В клинике ФМД проявляется развитием вазоренальной АГ в молодом возрасте. Дебют АГ приходится на 2-3 десятилетие жизни. Болеют чаще женщины, хотя описаны случаи и у мужчин. У больных с АГ проведение диагностического скрининга показано при
- Дебюте АГ до 30 лет, особенно у женщин
- Наличии АГ 3 ст. или злокачественная АГ
- Резистентной АГ на фоне комбинированной терапии 3 и более препаратами, включая диуретик
- Малом размере почек без анамнеза нефропатии
- Шуме над почечными артериями при отсутствии анамнеза атеросклероза
- ФМД других локализаций
Скрининговым методом обследования является проведение ЦДС артерий (особенно для почечных сосудов). Методами подтверждения диагноза – МСКТ или МРТ исследование, а также ангиография.
В лечении ФМД почечных сосудов методом выбора является проведение ангиопластики почечных сосудов.
Гиперурикемические тубулоинтерстициальные нефропатии
Три аутосомно-доминантные тубулоинтерстициальные нефропатии, такие как семейная ювенильная гиперурикемическая нефропатия, медуллярная кистозная болезнь почек и гломерулокистозная болезнь почек (уромодулиновая и уромодулино-подобная нефропатия) ассоциированы с мутациями в гене уромодулина. Эти заболевания характеризуются ювенильной гиперурикемией, подагрой и прогрессирующей почечной недостаточностью. Как правило, заболевание не сопровождается АГ на начальных стадиях, повышение АД возникает после начала формирования почечной недостаточности. Для всех заболеваний нехарактерным является выраженная протеинурия. Размер почек обычно уменьшается. Лекарственные препараты существенно не влияют на функцию почек и течение заболевания. Тяжелая почечная недостаточность, требующая проведения заместительной почечной терапии обычно развивается в возрасте 30-50 лет. На сегодняшний день в литературе содержится описание около 300 родословных.
| Название заболевания | Тип наследо-вания | № по класси-фикации Мак Кьюсика | Белок | Ген | Хромосомная локализация | Особенности клиники | Наличие подходов к лечению |
|---|---|---|---|---|---|---|---|
| Альдостеронизм, чувствительный к глюкокортикоидам | Аутосомно-доминантный | 103900 | 11β-гидроксилаза | Химерный ген CYP11B1, CYP11B2 | 8q21 | Уровень альдостерона нормально или умеренно повышен. Уровень КС и 17-ГКС – повышен. Ренин – низкий, калий – низкий. Диагностика – проба с дексаметазоном, обнаружение химерного гена | Лечение кортикостероидами |
| Синдром Лиддла (Псевдоальдостеронизм, тип I) | Аутосомно-доминантный | 177200 | Бета и гамма субъединицы эпителиального натриевого канала | SCNN1B SCNN1G |
16p13-p12 | Соль-чувствительная АГ, низкий калий Низкий ренин и альдостерон Начало АГ в детском или подростковом возрасте Диагностика – генетический анализ. |
Амилорид, триамтерен |
| Гипертония беременных (гипертония, индуцируемая прогестероном) Синдром Геллера | Аутосомно-доминантный | 605115 | Минералокортикоидный рецептор | NR3C2 | 4q31.1 | Тяжелая АГ и гипокалиемия развиваются во 2 и 3 триместре беременности. Повышение АД не сопровождается проявлениями эклампсии | Нет |
| Псевдоальдостеронизм типа II (синдром Гордона) | Аутосомно-доминантный | 145260 | Транспортер WNK | WNK1 WNK4 KLHL3 CUL3 |
1q31-q42 17q21 5q31 2q36 1q31-q42 |
Гиперкалиемия+метаболический ацидоз Нормальный уровень альдостерона Соль-чувствительность |
Хороший ответ на тиазидные диуретики |
| Синдром кажущегося избытка минералокортикоидов. | Аутосомно-рециссивный | 207765 | 11-бета-гидроксистероиддегидрогеназа | HSD11B2 | 16q22 | Повышенное АД, гипокалиемия, пониженные уровни ренина и альдостерона. Выраженный избыток метаболитов кортизола по сравнению с метаболитами кортизона в моче пациентов. |
Спиронолактон |
| Гиперурикемические тубулоинтерстициальные нефропатии | аутосомно-доминантные | 609886 603860 162000 |
Уромодулин | UMOD | 16p.12.3 | Гиперурикемия, гипертония, ХБП | Петлевые диуретики |
| Фибромускулярная дисплазия | Аутосомно-рециссивный | 608723 | фосфотазы и регулятора актина 1 типа | PHACTR1 | 6р.24.1 | Не атеросклеротический стеноз почечных артерий | Ангиопластика |
Гипертрофическая кардиомиопатия
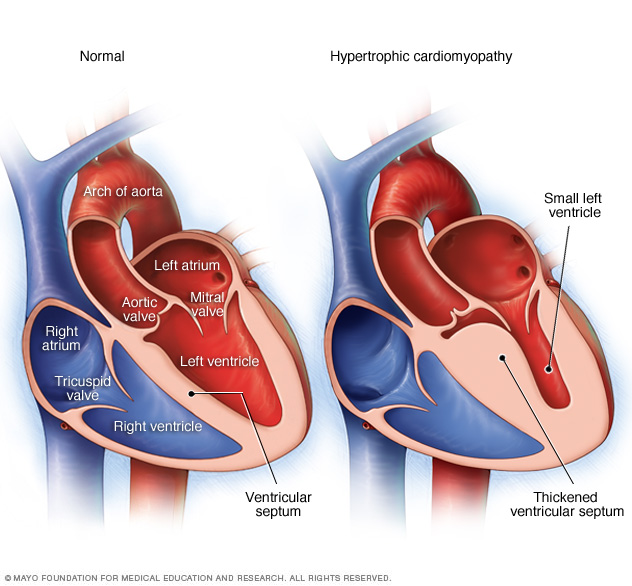
Гипертрофическая кардиомиопатия (ГКМП) – заболевание, характеризующееся утолщением стенок левого желудочка (ЛЖ), которое не может быть объяснено только нарушением внутрисердечной гемодинамики.
ГКМП – самое распространенное наследственное заболевание сердца. Больных с клиническими проявлениями в популяции 1:500, а если учесть и бессимптомных носителей мутаций 1:200.
У взрослых ГКМП подозревают при утолщении одного или более сегментов ЛЖ ≥ 15мм, определенном любым визуальным методом исследования (эхокардиография (ЭХОКГ), магнитно-резонансная томография или компьютерная томография). Диагноз ГКМП возможен и при меньшей толщине стенок ЛЖ (13-14мм), но при наличии других признаков болезни (семейный анамнез ГКМП, внекардиальные симптомы, изменения на электрокардиограмме (ЭКГ), отклонения от нормы в лабораторных показателях).
Клинические проявления ГКМП зависят от этиологии гипертрофии ЛЖ. Мутации в генах, кодирующих белки саркомера, являются основной причиной заболевания. Доля «саркомерной» ГКМП составляет около 60%. Остальные случаи кардиомиопатии представляют собой либо изолированное поражение сердца причиной которого стала мутация в гене не саркомера, либо гипертрофия ЛЖ является частью системного заболевания. При этом системное заболевание может быть, как наследственным, так и приобретенным. При системных болезнях, таким образом, у больных помимо гипертрофии ЛЖ будут и другие внекардиальные симптомы.
Клиническая манифестация ГКМП с изолированным поражением сердца может быть различной, и включает в себя признаки сердечной недостаточности (СН) (одышка), проявление обструкции выходного отдела ЛЖ (ВОЛЖ) (обмороки), боль в груди вследствие нарушения микроциркуляции в гипертрофированном миокарде ЛЖ, аритмии или внезапная сердечная смерть (ВСС).
Принимая во внимание гетерогенность этиологии ГКМП алгоритм постановки диагноза следующий: подозрение на ГКМП при визуальном методе исследования сердца (чаще ЭХОКГ), далее тщательный сбор семейного анамнеза в трех поколениях (случаи установленной ГКМП, ВСС в молодом возрасте) и оценка наличия внекардиальных симптомов системного заболевания. Подтверждением диагноза в большинстве случаев является результат генетического (мутация) либо лабораторного (например, обнаружение амилоида в тканевом биоптате при амилоидозе или дефицит альфа-галактозидазы в крови при болезни Фабри) анализа. Биопсия миокарда не показана для подтверждения диагноза ГКМП.
ГКМП чаще всего является наследственным моногенным заболеванием с преимущественно аутосомно-доминантным типом наследования. Генетический анализ у больных с клиническими признаками ГКМП оказывается положительным (присутствие мутации) в около 70% случаев, при этом 60% приходится на 9 основных генов саркомера, а на все остальные гены, которых установлено уже более 100, приходится суммарно <10%. Среди 30% больных с отрицательным генетическим анализом большинство относят также к наследственной ГКМП, но с неустановленной мутацией. И только около 5% носят приобретенный характер (амилоидоз, миокардит, феохромацитома, акромегалия, прием анаболических стероидов и прочее).
Особенностью ГКМП, как и других наследственных кардиомиопатий, является возраст-зависимая пенетрантность. Так, только 55% носителей патогенных мутаций имеют гипертрофию ЛЖ в возрасте 10-29 лет. Между 30 и 49 годами фенотипические проявления регистрируются у 75%, а в возрасте старше 50 лет у 95% носителей. Поэтому кардиологический скрининг (ЭКГ, ЭХОКГ) родственников пробанда первой линии в отсутствии возможности проведения генетического анализа рекомендовано начинать в возрасте 10-12 лет и заканчивать в 50-60 лет.
Больные с установленным диагнозом ГКМП с изолированным поражением сердца (чаще «саркомерная» ГКМП) должны подвергаться ежегодной оценке размеров толщины стенок ЛЖ, левого предсердия, обструкции ВОЛЖ. Также ежегодно рекомендовано проведение холтеровского мониторирования ЭКГ для выявления аритмий. Из жалоб особое внимание уделяется появлению обмороков и случаям ВСС среди молодых (до 40 лет) родственников. На основании этих данных при каждом посещении врача у больного с ГКМП рассчитывается 5-ти летний риск ВСС и принимается решение о необходимости имплантации кардиовертера-дефибриллятора. Это основная и единственная мера профилактики ВСС при ГКМП. Симптоматическое лечение СН зависит от морфологического варианта ГКМП (с обструкцией ВОЛЖ или без). Так, при обструктивной форме при градиенте давления в ВОЛЖ > 50ммрт.ст. показано хирургическое лечение (миосептэктомия, алкогольная аблация). В диапазоне 30-50ммрт.ст. при наличии жалоб (одышка, обмороки) применяют один из трех препаратов (бета-блокатор, верапамил или дизапирамид), способных снижать градиент давления в ВОЛЖ. В случае появления симптомов (одышка, боли в груди) у больных без обструкции ВОЛЖ используется стандартная схема лечения СН. В ряде случаев в качестве лечебного мероприятия прибегают к протезированию митрального клапана, так как у около 20% больных с ГКМП регистрируется сопутствующая патология передней створки МК, усугубляющая обструкции в ВОЛЖ и приводящая к выраженной митральной недостаточности. Генетическое консультирование и генетический анализ показан всем больным с гипертрофией миокарда ЛЖ ≥ 15мм, необъяснимой только лишь нарушениями пред- или постнагрузки (класс доказанности IB). И наконец, при появлении фибрилляции предсердий у больного с ГКМП следует сразу назначить пероральный антикоагулянт без оценки показаний по шкале CHA2DS2-VASc вследствие высокого риска инсульта.
Тромбофилия
Тромбофилия – это нарушения гемостаза и гемореологии, характеризующиеся повышенной склонностью к развитию тромбозов или внутрисосудистого свертывания, в основе которых лежат приобретенные и генетически обусловленные нарушения. Генетически обусловленные нарушения в системе гемостаза определяют термином «наследственная тромбофилия». Наиболее изучено влияние наследственной тромбофилии на риск развития венозных тромбоэмболических осложнений, включающих тромбоз глубоких вен и тромбоэмболию легочной артерии, а также на риск развития редких видов венозных тромбозов. Ряд авторов расширяют понятие наследственной тромбофилии в аспекте предрасположенности к артериальным тромбозам в молодом возрасте.
Современные клинические рекомендации обсуждают целесообразность выявления в различных ситуация только факторов «классической» тромбофилии - дефицит антитромбина, дефицит протеинов С и S, лейденскую мутацию и мутацию G20210A гена протромбина. В настоящее время для выявления дефицита естественных антикоагулянтов (антитромбина, протеинов С и S) используются иммунологические и коагулогические методы, что существенно ограничивает возможности тестирования пациентов в остром периоде тромбоза, во время беременности или на фоне лечения антикоагулянтами. Использование методов полноэкзонного секвенирования способно повысить выявляемость тромбофилии, ассоциированной с дефицитом естественных антикоагулянтов, и снизить стоимость тестирования.
«Классические» наследственные тромбофилии встречаются достаточно редко и не способны объяснить развитие тромбоэмболических осложнений в большинстве случаев. Предполагается, что значительная часть больных с тромботическими осложнениями являются носителями других генетических вариантов, не входящих в стандартную панель тестирования «классической тромбофилии». Внедрение методов полногеномного поиска ассоциаций и секвенирования нового поколения позволило разработать новую панель, содержащую 56 генетических маркеров, ассоциированных с риском развития артериальных и венозных тромбозов.
Проведение генетического анализа для выявления тромбофилии целесообразно у женщин, планирующих беременность или прием гормональных контрацептивов, у лиц с редкими типами венозных тромбозов (тромбоз венозного синуса, неспровоцированный тромбоз вен верхних конечностей, тромбоз абдоминальных вен и пр.), у пациентов, перенесших эпизод тромбоза глубоких вен или тромбоэмболию легочной артерии (в спорных случаях, для решения вопроса о пролонгировании антикоагулянтной терапии) и у пациентов с парадоксальными артериальными эмболиями или при раннем дебюте артериального тромбоза (до 50 лет) в отсутствии традиционных факторов риска.